driftR: an interactive population genetic simulation website that allows students to explore the impacts of genetic drift, selection, migration, mutation, and population sizes on a variety of summary statistics.
https://cjbattey.shinyapps.io/driftRLDsim: simulate and plot visualizations of pairs of alleles in a small population. Designed for teaching intro lessons about linkage and linkage disequilibrium.
https://cjbattey.shinyapps.io/LDsimadaptR: simulate selective sweeps and other processes with varying selection over time.
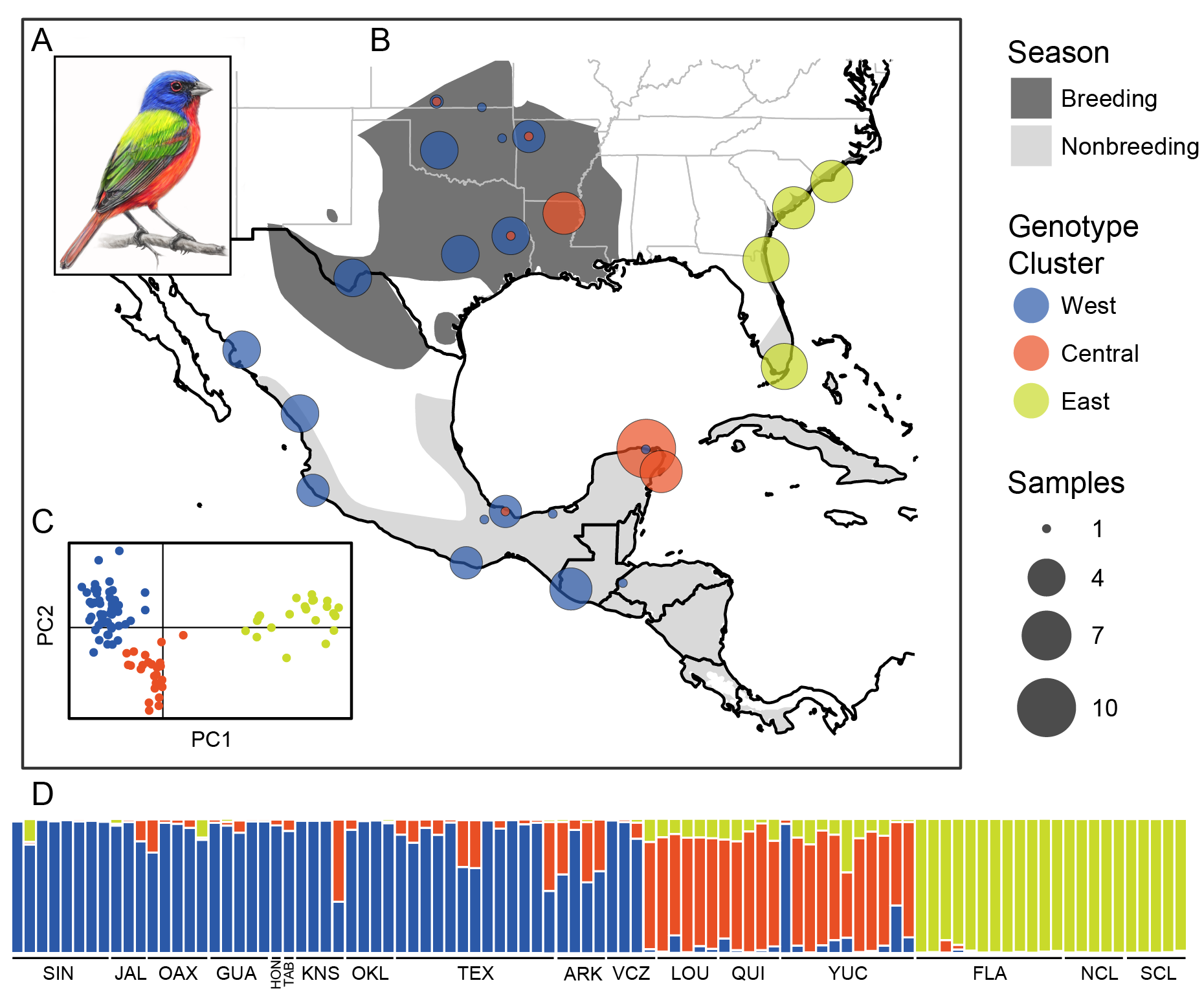
https://cjbattey.shinyapps.io/adaptRstructurePlotter: plot output of genotype clustering algorithms with fancy color selection and a permutation algorithm to deal with label switching.
https://cjbattey.shinyapps.io/structurePlotter
Sanderlings. Florence, Oregon.

Ship Rock. Oregon.

Anna's hummingbird. Seattle, Washington.

Ballard, Seattle, Washington.

Anna's hummingbird. Seattle, Washington.

Cedar Waxwing. Lolo NF, Idaho.

Blue-throated Hummingbird. Nuevo León, MX.

Anna's Hummingbird. Seattle, WA.

Over Cerro Catedral. Nahuel Huapi, Argentina National Park

Mountain Goat. Olympic National Park, WA.

Short-eared owl lost her vole to a Harrier. Skagit Valley, WA.

Columbia spotted frog. Swan River, MT.

Cottontail. Estes Park, CO.

Mountain Bluebird. Estes Park, CO.

Brown-backed Solitaire. Nuevo León, MX.

California newts (Taricha torosa) breeding in the Big Sur River. Monterey County, California

Stillwater Canyon, UT.
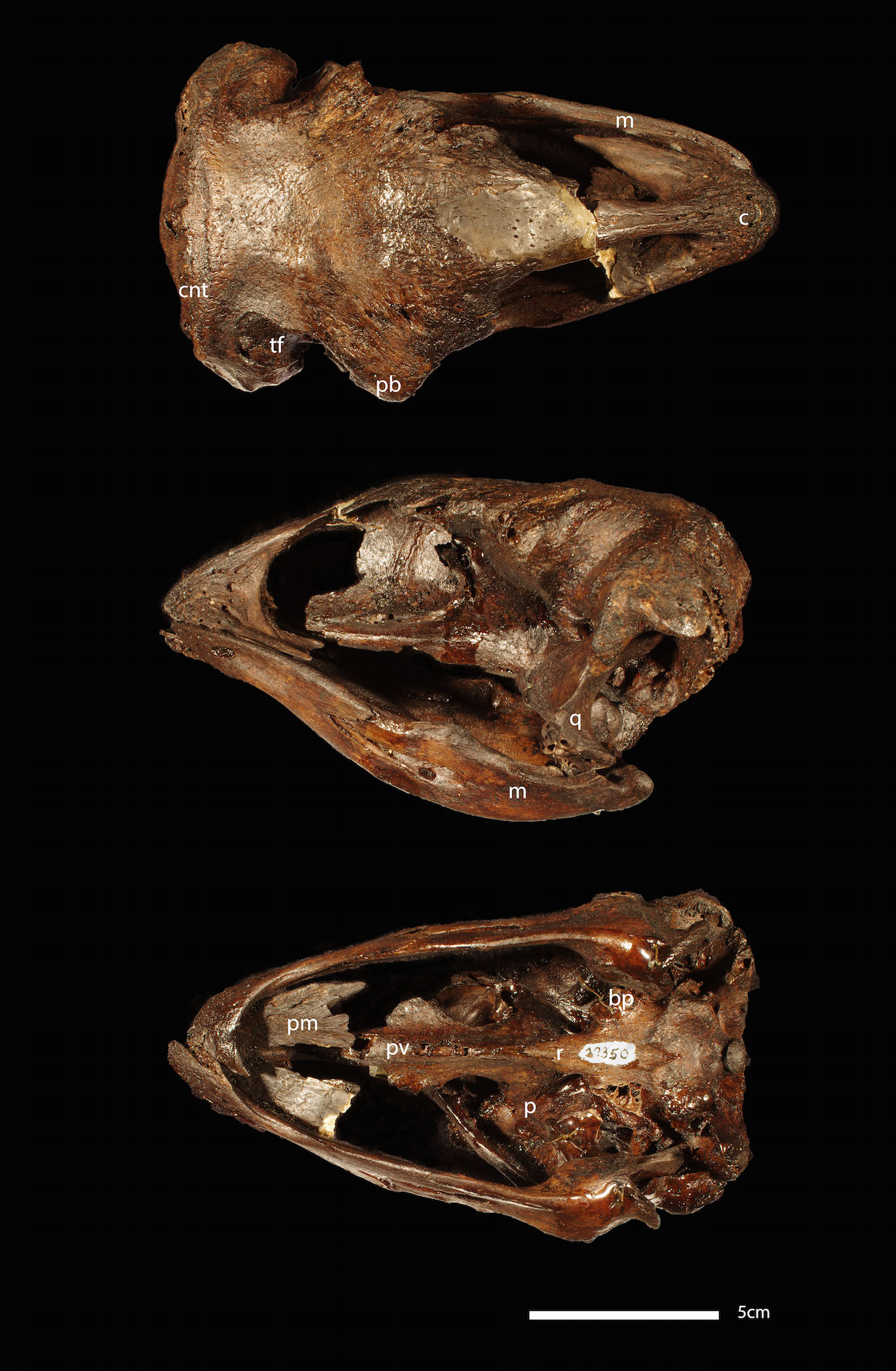
Moa skull. Burke Museum of Natural History, Seattle.

Labyrinth Canyon, UT.

Harlequin Ducks. Olympic National Park, WA.

Macgillivray's warbler (Geothlypis tolmiei). Pescadero, California

American Kestrel chicks. Hopland, CA.

Weighing a bunting. Pescadero, CA.

Lazuli Bunting. Pescadero, CA.

Blue-naped Chlorophonia. Santa Marta Mtns, Colombia.

American Kestrel. Blue Oak Reserve, CA.

Lazuline Saberwing. Santa Marta Mtns, Colombia.

Violet-crowned Woodnymph. Santa Marta Mtns, Colombia.

California red-legged frog. Pinnacles National Park, CA.

California Newts. Big Sur River, CA.

Taking pictures like this used to be my job. Mendocino County, CA.

Allen's hummingbird (Selaslphorus sasin). Pescadero, California

weighing a lazuli bunting (Passerina amoena). Pescadero, California

American kestrel (Falco sparverius) chicks. Hopland, California

American kestrel (Falco sparverius) chick. Hopland, California
cjbattey@gmail.com
CJ Battey
Computational Biologist
Myriad Genetics
San Francisco, California